
© Olivier Le Queinec, 123RF.com
Das Chemical Development Kit bereichert die Chemoinformatik um eine solide Bibliothek und hilft dabei, herkömmliche Anwendungen für den Einsatz in der Naturwissenschaft fit zu machen. Die Ergebnisse kommen beispielsweise der Forschung an Medikamenten gegen Malaria zugute.
Das Konzept der freien Software entstammt dem akademischen Bereich und viele freie Projekte entstehen in einem wissenschaftsnahen Umfeld. Dennoch ist es eine verhältnismäßig neue Entwicklung, dass die Konzepte von Open Source die Art und Weise beeinflussen, in der naturwissenschaftliche Forschung betrieben wird. Open Data oder Open Notebook Science [1] sind Ansätze, die sich explizit auf die offene Software-Entwicklung beziehen (siehe Kasten “Glossar”).
|
Glossar |
|---|
|
Blue Obelisk: Gruppe von Chemikern und Programmierern, die sich für offene Standards, Open Data und Open Source in der Chemie einsetzt. CML: Ein XML-basiertes Dokumentenformat zur Darstellung chemischer Formeln und anderer Sachverhalte. Die Entwickler bezeichnen es als “HTML für Moleküle”. Inchi: Eine von der IUPAC (International Union of Pure and Applied Chemistry, der wichtigste Herausgeber von Standards in der Chemie) standardisierte Notation von chemischen Strukturformeln als Zeichenketten. NMR (Kernmagnetische Resonanzspektroskopie): Verbreitete Methode zur Strukturaufklärung insbesondere organischer Moleküle. NMR beruht darauf, dass manche Atomkerne einen Spin, also vereinfacht ein magnetisches Drehmoment haben. Deshalb lassen sich Wechselwirkungen mit einem angelegten magnetischen Feld beobachten. Die Wechselwirkungen einzelner Atomkerne sind abhängig von der “elektronischen Umgebung” des Kerns, also von der ihn umgebenden Elektronenhülle, und anderen “magnetischen” Kernen in der Nähe. Daraus lassen sich Strukturinformationen gewinnen. Open Notebook Science: Ein von Open Source inspiriertes Prinzip der naturwissenschaftlichen Forschung, bei der Wissenschaftler Versuchsabläufe und Rohdaten von Ergebnissen zeitnah für alle einsehbar ins Internet stellen, das Laborjournal (Notebook) der Forscher also offen zugänglich ist. Pubc Chem: Ein Verbund frei zugänglicher Datenbanken mit Millionen von chemischen Strukturformeln und anderen Informationen über Moleküle. QSAR/QSPR: QSAR (Quantitative Structure Activity Relationship) und QSPR (Quantitative Structure Property Relationship) stellen Beziehungen zwischen der Struktur eines Moleküls und seinen Wirkungen beziehungsweise Eigenschaften dar. Das können pharmakologische oder biologische Wirkungen sein, wie sie zum Beispiel in der Pharmaforschung wichtig sind, aber auch physikalische oder chemische Eigenschaften. Oft wird der Begriff QSAR sowohl für biochemische Wirkungen als auch physikalische Eigenschaften verwendet. Ein sehr einfaches Beispiel für QSPR stellt der Zusammenhang zwischen der Kettenlänge ansonsten gleich aufgebauter (homologer) Kohlenwasserstoffe und deren Siedepunkt dar, ein etwas praxisnäheres wäre die Wirkung eines Moleküls auf ein bestimmtes Enzym. Dem Molekül oder einzelnen seiner Atome ordnet man in einer Reihe so genannter Deskriptoren physikalische, chemische, topologische oder elektrische Eigenschaften zu und ermittelt dann Korrelationen zu Wirkungen. QSARs für die pharmakologischen Forschung herauszufinden ist ein komplizierter Vorgang und erfordert Methoden zur Hypothesenbildung sowie aufwändige statistische Berechnungen. Smiles (Simplified Molecular Input Line Entry System): Verbreitete Notation, um chemische Strukturformeln vorzugsweise organischer Moleküle als Ascii-Strings zu repräsentieren. Das Format wird von der Firma Daylight kontrolliert, die offene Variante Open Smiles stammt von der Vereinigung Blue Obelisk. |
Modular und interoperabel
In diesem Umfeld entsteht wiederum Open-Source-Software, die den Anforderungen entspricht, die diese Prinzipien stellen: modular, interoperabel, Web-zentriert. Ein Beispiel hierfür bieten das Chemical Development Kit (CDK, [2]) und die Software, die Forscher mit dessen Hilfe entwickeln. Dieser Artikel soll einen kleinen Einblick in dieses Umfeld bieten, mehr jedoch nicht, denn das CDK erfordert sehr viel Einarbeitungszeit. Professor Zielesny von der FH Gelsenkirchen, dessen Arbeitsgruppe das Development-Kit intensiv nutzt, rechnet damit, dass ein begabter Chemiestudent mit viel Interesse an Programmierung etwa drei Monate benötigt, bis er brauchbare Ergebnisse damit produziert.
Als das Linux-Magazin im Jahre 2001 das Projekt zum ersten Mal kurz vorstellte [3], war es gerade gestartet und der Erfolg nicht absehbar. Acht Jahre später gibt es ein gutes Dutzend Anwendungen, die das Toolkit nutzen und eine höchst aktive Entwicklergemeinde (siehe Kasten “Interview mit Egon Willighagen”). Auf dem CDK-Workshop in Cambrigde trafen sich im April über 40 Teilnehmer, um über die Entwicklung und Anwendung des CDK zu diskutieren.
|
Interview mit Egon Willighagen, |
|---|
|
Linux-Magazin: Wenn Sie den heutigen Stand des CDK mit dem vergleichen, was Sie vorhatten, als das Projekt startete, wie hat es sich entwickelt? E.-Willighagen: Das CDK ist viel größer geworden, als ich damals geglaubt hatte. Der Artikel von damals wurde inzwischen 66-mal zitiert und wir haben mehr als 50 Leute, die Beiträge geleistet haben, auch wenn viele davon eher klein waren. Das CDK mag zwar nicht immer so mächtig sein wie kommerzielle Tools, aber wir haben eine Bibliothek mit allen Grundfunktionen und ein paar ausgesprochenen Perlen hier und da. Die offene Natur des Projekts gibt den Nutzern das Vertrauen, das sie vielleicht auch kommerziellen Angeboten entgegenbringen. Linux-Magazin: In vielen Ihrer Publikationen setzen Sie sich für Open-Source-Chemoinformatik ein, weil sowohl die Algorithmen als auch die Implementierung einer Peer Review unterzogen werden kann. E.-Willighagen: Es gibt natürlich Ansätze, die Peer Reviews von proprietärer Software erlauben, aber niemals in der Transparenz wie bei Open Source. Kommerzielle Lizenzen haben oft Klauseln, die bestimmte Dinge verbieten, zum Beispiel konkurrierende Implementierungen zu bauen. Damit verhindert man vernünftige Evaluierung. Linux-Magazin: Warum wird Open-Source-Software für ernsthafte wissenschaftliche Arbeit immer noch so selten in großem Stil eingesetzt? E.-Willighagen: Ich glaube, das liegt daran, wie Wissenschaft finanziert wird. Die Forschung erledigen zum großen Teil Doktoranden und Postdocs mit Zeitverträgen. Wenn diese beendet sind, hören sie auf, sich um die Software zu kümmern. Für mich war Open-Source-Chemoinformatik immer ein Hobby und hatte zum Beispiel mit meiner Doktorarbeit nichts zu tun. Die meisten anderen Projekte in der Chemoinformatik sind ziemlich klein, CDK ist eines der größten. Linux-Magazin: Wie finanziert sich die Entwicklungsarbeit? E.-Willighagen: Nicht direkt. Teile der Forschungsarbeit sind oft wieder verwendbar und finden dann ihren Weg in Softwarebibliotheken. Das CDK wird somit also indirekt mitfinanziert. Die meiste Zeit als Release Manager hatte mein normaler Job nichts mit dem CDK zu tun. Christoph Steinbeck ist in der glücklichen Lage, Leute für Forschung in der Chemoinformatik bezahlen zu können, und er glaubt wie ich an Open Source. Linux-Magazin: Was könnte man tun, damit die Entwicklung nachhaltiger wird? E.-Willighagen: Christoph Steinbeck [Mitinitiator des CDK, Anm. d. Red.] hat jetzt durch das Industry Program am European Bioinformatics Institute wieder ausgezeichnete Kontakte zur Industrie, die großes Interesse am CDK zeigt. Ich selbst habe mich der Bioclipse-Gruppe angeschlossen und sehe das auch als wichtige Plattform, um das CDK zu promoten. Bei Bioclipse könnte möglicherweise ein kommerzielles Spin-off entstehen, das dann auch die Entwicklung von Open-Source-Software finanzieren sollte. Was die größere Verbreitung angeht, kann man die Rolle der Linux-Distributionen kaum überbewerten, vor allem die Standardisierungen im Paketmanagement, beispielsweise bei Debian GNU/Linux. Jedes vernünftige freie Projekt sollte seine Software für die Distributionen verfügbar machen, also Portabilität und offene Standards beachten. Linux-Magazin: Zur Beliebtheit einer Software trägt ja auch gute Dokumentation bei. Können wir bald bessere Dokumentation oder Tutorials zum CDK erwarten? E.-Willighagen: Die CDK-Entwickler werden nur dafür bezahlt, die Software bei der Forschung einzusetzen. Als Folge davon leidet die Dokumentation, weil niemand einen Grund sieht, sich wirklich darum zu kümmern. Aber sie wird sich verbessern. Ich arbeite gerade an einem Buch “Groovy Cheminformatics” mit vielen Codebeispielen zum CDK mit Groovy. Davon wird einiges als Dokumentation ins Projekt zurückfließen. Linux-Magazin: Letzte Frage, was macht bei der Arbeit am CDK am meisten Spaß? E.-Willighagen: Die Erfahrung, dass es Leuten tatsächlich dabei hilft, ihre Forschungsarbeit zu erledigen. Linux-Magazin: Und was nervt gelegentlich? E.-Willighagen: Die fehlende Zeit, um mehr Programmcode zu schreiben und diesen auch besser zu dokumentieren. |

Das unter der LGPL lizenzierte CDK ist zunächst einmal eine Ansammlung von Java-Klassen, die Methoden der Chemo- und Bioinformatik bereitstellt. Damit lassen sich einerseits Anwendungen für spezifische Forschungsprojekte schreiben, die Arbeitsgruppen zur Lösung chemoinformatischer Probleme einsetzen. Viel spannender ist aber, dass es das CDK erlaubt, bestehende Anwendungen und Frameworks in Werkzeuge für Chemiker, Biologen und Pharmakologen zu verwandeln. So wurde Eclipse zu Bioclipse und das freie Workflowsystem Taverna verwandelte sich in CDK-Taverna.
|
Erste Schritte mit dem CDK und |
|---|
|
Das folgende Beispiel setzt voraus, dass auf Ihrem System eine lauffähige Java-Implementierung existiert, beispielsweise JDK 1.6.x oder zumindest eine JRE. Als Umgebung, um das CDK auszuprobieren und einige Funktionen zu demonstrieren, eignet sich die Skriptsprache Groovy, weil sie auch ohne Java-Kenntnisse nahezu intuitiv einsetzbar ist und deutlich weniger Code erfordert als Java. Zur Installation von Groovy im Homedirectory folgen Sie den knappen Anweisungen auf [14]. Das dort angebotene Zip-Paket sollte auf allen aktuellen Linux-Distributionen einsetzbar sein. Besorgen Sie sich dann das Jar-File mit dem aktuellen CDK von [2] (beispielsweise »cdk-1.2.2.jar«) und fügen dessen Pfad zum Classpath hinzu. Alternativ können Sie die Datei auch direkt in das Verzeichnis »lib« unterhalb der Groovy-Installation kopieren. Starten Sie dann mit ./Groovy-Verzeichnis/bin/groovyConsole die Groovy-Konsole. Dort lassen sich Groovy-Dateien öffnen oder kleine Codeschnipsel direkt eingeben und mit [Strg]+[R] ausführen. Die Groovy-Konsole enthält auch einen Objektbrowser, der ganz nützlich ist, um sich etwas in den Klassen des CDK umzuschauen (»Script | Inspect Last« ). Das folgende Listing erzeugt zunächst einmal etwas (virtuellen) Ethylalkohol und demonstriert damit das Anlegen von Atomen und Bindungen.
import org.openscience.cdk.interfaces.*
import org.openscience.cdk.*;
atom1 = new Atom("C")
atom2 = new Atom("C")
atom3 = new Atom("O")
bond1 = new Bond(atom1, atom2, IBond.Order.SINGLE)
bond2 = new Bond(atom2, atom3, IBond.Order.SINGLE)
Viele weitere Beispiele, um mit Groovy das CDK zu entdecken, finden sich in [15]. |
Das CDK baut also eine Brücke, die die Welt von Chemikern und Biologen mit modernen Entwicklungen in der IT-Welt verknüpft, zum Beispiel mit Webservices und Distributed Computing, datengetriebenen Anwendungen und agilen Entwicklungsmethoden.
Chemo-Java
Das CDK definiert zunächst einige grundlegende Klassen und Methoden, um Atome zu Molekülen zu verbinden und physikalisch-chemische Eigenschaften zuzuordnen (siehe Kasten “Erste Schritte mit dem CDK und Groovy”). Eine weitere wichtige Gruppe sind I/O-bezogene Klassen. Ein Chemiker wird sich in den seltensten Fällen seine Molekülrepräsentationen selbst zusammenbasteln, sondern auf Datenbanken wie etwa Pubchem [4] zurückgreifen.
Das CDK versteht eine Vielzahl von Formaten, etwa Smiles, Inchi oder CML, und implementiert Methoden, um sich Daten aus den Strukturdatenbanken zu holen und die Ausgabe so zu gestalten, dass sie von 3D-Renderern wie Jmol oder anderer Software verstanden wird.
Weitere Klassen implementieren Schnittstellen für QSAR-Berechnungen [5], Voraussagen von NMR-Spektren und vieles mehr. Umfassende Informationen liefert das Javadoc-Archiv, das auf der Projekt-Website [2] erhältlich ist, einen brauchbaren Überblick gibt [6].
Bioclipse
In Bioclipse, einer Eclipse-basierten Arbeitsplattform für Bioinformatiker, Biologen und Chemoinformatiker hat sich ein CDK-Plugin [7] mittlerweile einen festen Platz erobert (Abbildung 1). Doch nicht nur die Java-Welt profitiert von der Bibliothek. Recht weit gediehen ist das Cinfony-Projekt [8], das Python-Schnittstellen zu CDK und anderen Chemoinformatik-Werkzeugen bereitstellt. Ruby-CDK [9] war einige Zeit verwaist, hat aber zwei Tage vor Redaktionsschluss mit einem neuen Maintainer wieder Fahrt aufgenommen. Komplett sprachneutral wird der Zugriff auf die Software, wenn sie als REST-basierter Webservice implementiert ist [10].
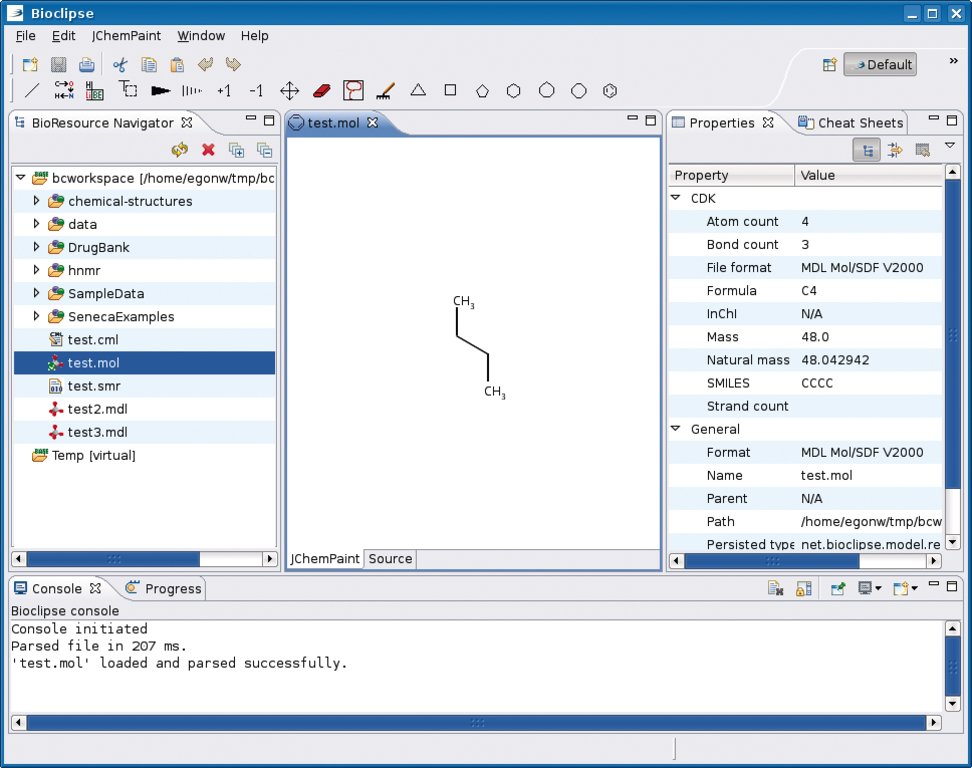
Abbildung 1: Das CDK-Plugin in Bioclipse bei der Darstellung von Butan.
CDK-Taverna
Ein gutes Beispiel, wie das CDK seinen Weg in andere Projekte findet, ist die Integration in das Workflow-System Taverna. Workflow-Systeme dienen dazu, Arbeitsabläufe ganzer Organisationen, Arbeitsgruppen oder Einzelpersonen zu formalisieren. Wichtige Ziele sind, wiederkehrende Fehler zu vermeiden, die Abläufe definiert zu dokumentieren und aus vorhandenen Abläufen neue zu erzeugen. In Unternehmen kommen verschiedene Workflow-Systeme zum Festlegen der Geschäftslogik zum Einsatz.
Doch besonders dann, wenn es um die Verarbeitung großer Datenmengen aus unterschiedlichen Quellen und in vielen verschiedenen Formaten geht, sind Workflow-Systeme auch für die Wissenschaft interessant. Taverna [11] ist eine Software, die ein Team des European Bioinformatics Institute (EBI) und mehrerer britischer Universitäten speziell für diesen Anwenderkreis entwickelt hat. Ursprünglich für die Bioinformatik gedacht, finden sich inzwischen auch Anwender aus den Gesellschaftswissenschaften und der Chemoinformatik.
Workflows lassen sich in Taverna mit einer grafischen Oberfläche, der Workbench, definieren (Abbildung 2). Die Abarbeitung des Workflow, also die Verarbeitung der Daten, obliegt einer Vielzahl so genannter Prozessoren, deren Zusammenwirken von einer Zwischenschicht mittels eines SCUFL-Modells gesteuert wird. SCUFL steht für Simple Conceptual Unified Flow Language, einer XML-basierten Sprache, um Workflows zu repräsentieren.
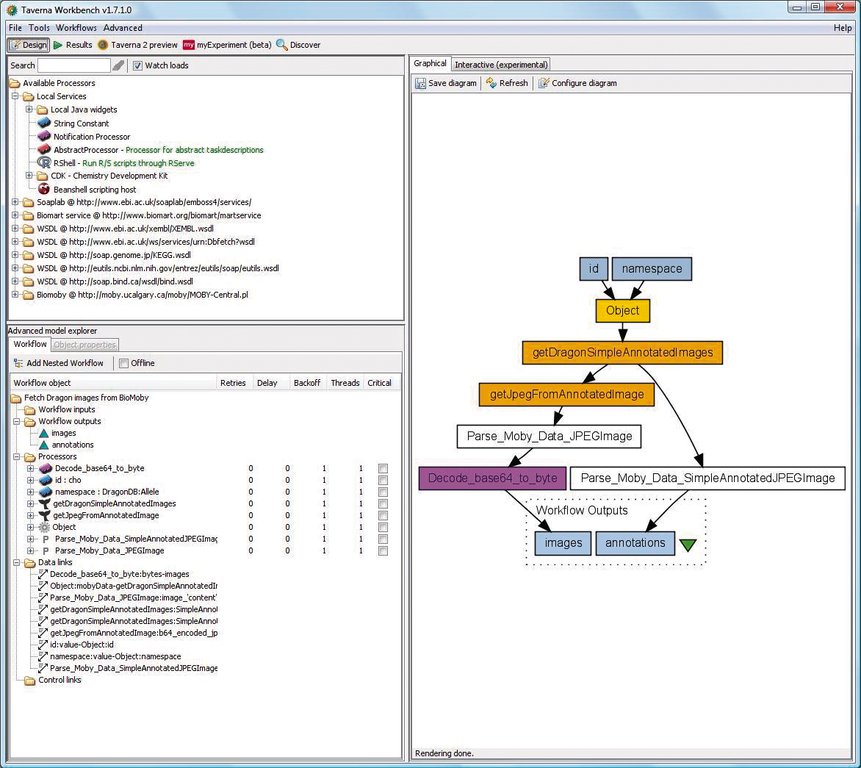
Abbildung 2: Das grafische Programm Taverna Workbench dient zur Modellierung von Workflows. Hier entsteht ein kleiner Arbeitsablauf, der Pflanzenbilder und Anmerkungen aus der Datenbank Biomoby holt.
Wer sich für die – recht komplexen – Details und Hintergründe von Taverna interessiert, kann außer auf der Projekt-Website auch aus Thomas Kuhns Dissertation [12] an der Uni Köln Näheres erfahren. Sie beschreibt CDK-Taverna, ein Plugin, dass es ermöglicht, mit Taverna Probleme der Chemoinformatik zu bearbeiten. Thomas Kuhn hat dafür etwa 160 Prozessoren geschrieben, die CDK-Methoden nutzen, um Moleküldaten aus Datenbanken zu holen, QSAR-Methoden darauf anzuwenden und die Ergebnisdaten zu präsentieren. Damit hat er unter anderem eine Diversitätsanalyse an über 200000 Molekülen durchgeführt, also eine automatische Klassifizierung von Substanzen nach bestimmten – in diesem Fall molekularen – Eigenschaften.
CDK-Taverna macht nach Abschluss dieser Dissertation einen etwas verwaisten Eindruck. Nach Auskunft von Professor Achim Zielesny von der FH Gelsenkirchen, einem der Initiatoren des Projekts, soll die Entwicklung demnächst weitergehen, zunächst mit einer Portierung auf Taverna 2. Immerhin gebe es bereits einige Anwender der Software im Bereich der Forschung und der Industrie.
Chemie-Experimente im Web 2.0
Eng verbunden mit Taverna ist die Website Myexperiment.org [13] von der Universität Manchester. Myexperiment ist eine Ruby-on-Rails-basierte Plattform, die im typischen Web-2.0-Aufzug daherkommt. Hier können Wissenschaftler Taverna-Workflows einstellen und miteinander teilen. Darunter sind auch einige CDK-Taverna-Workflows.
Zu den Anwendern des CDK gehört auch das Projekt Useful Chem. Neben anderer freier Software kommt dort das CDK für QSAR-Modellierungen zum Einsatz. Useful Chem ist ein interuniversitäres Projekt, dass sich dem Ansatz der Open Notebook Science verschrieben hat, und deren Grundsätze dazu verwendet, neue Malaria-Medikamente zu entwickeln.
Initiator von Useful Chem ist Professor Jean-Claude Bradley von der Drexel University in Philadelphia, dessen Arbeitsgruppe eng mit Rajarshi Guha von der Universität Indiana zusammenarbeitet und mit zahlreichen anderen Forschern, etwa beim EBI, kooperiert.
Laut Bradley führt das Prinzip der Open Notebook Science, Ergebnisse von Experimenten sofort ins Netz zu stellen, dazu, dass Forschungspartnerschaften ad hoc entstehen, wenn für bestimmte Fragestellungen woanders Lösungsansätze bereits existieren.
Useful Chem ist sehr offen, wenn es darum geht, neue Phänomene im Internet für Forschung zu nutzen – oder zu zweckentfremden. So hat sich Second Life zu einer beliebten Plattform für animierte 3-D-Darstellungen von Molekülen und Reaktionsabläufen entwickelt. (Abbildung 3, [16])
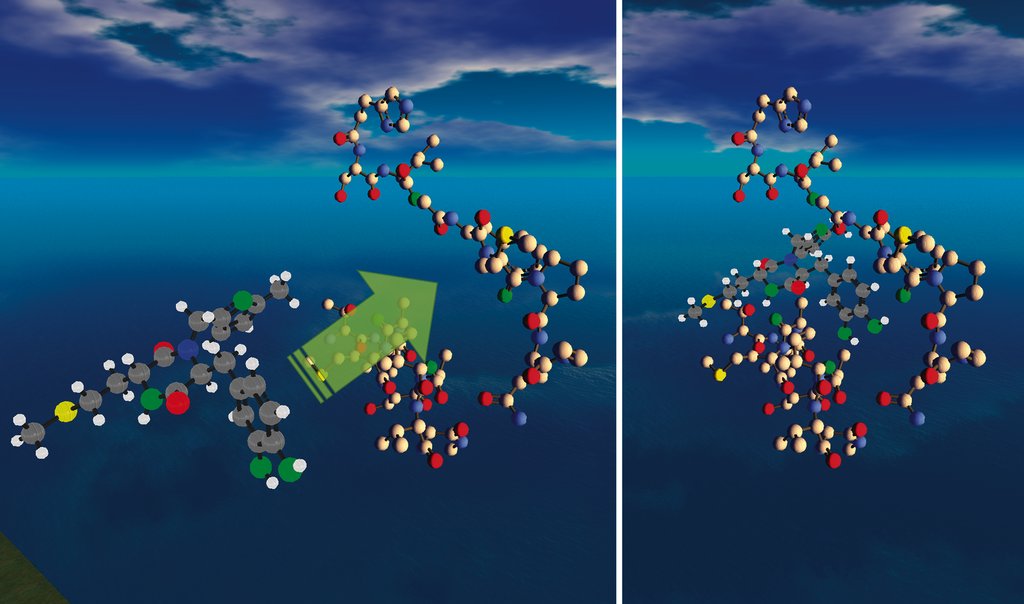
Abbildung 3: Second Life im Dienst der Wissenschaft: Ein Ugi-Produkt dockt an das Enzym Enoyl-Reduktase an. So dient das virtuelle Spiel-Universum ernsthafter Pharma-Forschung.
Wirkstoff gesucht
Malaria-Forschung ist für das Open-Notebook-Prinzip besonders geeignet, weil das Interesse großer Pharmakonzerne nicht sehr ausgeprägt ist. Die Erkrankten in Entwicklungsländern sind schlicht nicht zahlungskräftig genug. Useful Chem hat das Ziel, nach besonders billig herzustellenden neuen Wirkstoffen zu suchen, und forscht dabei an einer Substanzklasse, die zwei Enyzme des Malaria-Erregers, die Enoyl-Reduktase und Falcipain-2, blockieren kann. Ausgangspunkt war die schon länger bekannte Tatsache, dass bestimmte ringförmige Moleküle, die Diketopiperazine, diese Wirkung haben. Solche Substanzen ließen sich aber nur aufwändig aus Pilzen extrahieren oder in einer komplizierten Feststoffreaktion herstellen.
Bradleys Gruppe fand heraus, dass sie sich auch auf einem einfacheren Weg gewinnen lassen, der über die so genannte Ugi-Reaktion in organischen Lösungsmitteln führt. Diese liefert Zwischenprodukte, die noch nicht ringförmig sind.
Um Diketopiperazine herzustellen, wäre also ein weiterer Reaktionsschritt erforderlich, der sich als schwierig herausstellte. Chemoinformatische Modellierungen liefern jedoch Hinweise darauf, dass auch die Zwischenprodukte schon an das Enzym andocken können, sodass dieser Reaktionsschritt entfallen kann.
Praktischer Erfolg
Im Experiment hat sich tatsächlich eine Wirkung gezeigt, die zwar schwächer war als erwartet, aber dennoch weitere Untersuchungen in dieser Richtung rechtfertigt. Jetzt geht es der Arbeitsgruppe vor allem darum, die Ugi-Reaktion so zu optimieren, dass die Produkte sofort in fester Form anfallen. Da immer an ganzen Substanzklassen ähnlicher Produkte, so genannten Libraries, geforscht wird, es also sehr viele mögliche Endprodukte und Ausgangsstoffe gibt, ist es nötig, Voraussagen über die Löslichkeiten zu treffen. Experimentelle Daten existieren kaum, also ergibt sich ein typisches QSAR-Problem. Mit Hilfe des CDK und der freien Statistiksoftware R [17] sucht Rajarshi Guha an der Universität Indiana zusammen mit anderen Wissenschaftlern auf chemoinformatischem Weg nach dessen Lösung (mhu).
|
Der Autor |
|---|
|
Ulrich Wolf ist Diplomchemiker, er arbeitet als freier Journalist und berät Unternehmen beim Einsatz von Open-Source-Software. |





