
Bei der Suche nach neuen Medikamenten ziehen Forscher auch die Erbinformationen von Menschen und Tieren als Informationsquelle heran. Ein Open-Source-Projekt leistet mit seiner Genom-Datenbank bei der Suche nach Krankheitsursachen und dem Verständnis von Krankheitsverläufen wertvolle Hilfe.
Die Suche nach neuen Medikamenten hat viele Gesichter. Eines der häufig verwendeten Verfahren klingt sehr simpel: Die Forscher tropfen in langen Versuchsreihen chemische Substanzen auf Zellhaufen, die im Reagenzglas gezüchtet wurden, und beobachten die Wirkung. Tritt eine solche auf, dann gab es eine Wechselwirkung von Proteinen (oder Eiweißen) der Zellen mit dem potenziellen Medikament. Damit das Ganze nicht völlig planlos geschieht, gibt es die Bioinformatik. Sie speichert das Wissen über bereits bekannte Proteine, sagt die Existenz neuer Proteine voraus und wagt sich sogar an die Bestimmung ihrer Eigenschaften.
Proteine sind die Produkte unserer Gene. Sie werden in der DNA als Sequenz von vier verschiedenen Nucleinsäuren in der Zelle kodiert, meist kurz als A, C, G und T abgekürzt. Die Bestimmung dieser Reihenfolge ist die Sequenzierung des Genoms, die immer wieder für Medienschlagzeilen gut ist. Seit dem 14. April dieses Jahres ist die Sequenzierung des menschlichen Genoms nun offiziell abgeschlossen.
Sequenziert wurde es gleich zweimal. Von der amerikanische Firma Celera, die ihre Daten jetzt kommerziell vertreibt, und von einem internationalen Verbund akademischer Sequenzierzentren. Den europäische Anteil an der Sequenzierung des menschlichen Genoms hat größtenteils das Sanger Center[6] bei Cambridge geleistet, finanziert durch den Wellcome Trust.
Freie Software für frei zugängliche Daten
So offen wie die aus dem Sequenzier-Projekt gewonnenen Daten ist auch die Software, die die Unmengen an gewonnenen Daten verwaltet, zusammenfügt und präsentiert: das vom Sanger Center zusammen mit dem europäischen Bioinformatik-Institut (EMBL-EBI,[2], [3]) entwickelte Open-Source-Projekt Ensembl[1]. Es ist implementiert mit Apache, MySQL, Perl und zunehmend auch mit Java. Der aktuelle Entwicklungsstand ist über CVS erhältlich, Dumps der Datenbank über FTP.
Derzeit gibt es dort außer dem menschlichen Genom die Daten des Zebrafischs, der Maus, der Ratte und des Moskitos. Durch eine Kooperation mit Singapur kam auch der genetische Code des fernöstlichen Kugelfischs Fugu auf die Ensembl-Website. Das sind die Genome höherer Organismen, deren Sequenzierung am weitesten fortgeschritten ist. Sie wurden wegen ihrer großen Bedeutung in den Labors gewählt: zum besseren Verstehen einer bestimmten Krankheit (Moskito als Träger des ebenfalls sequenzierten Malaria-Erregers Plasmodium falciparum) oder beim Kugelfisch wegen außergewöhnlicher zellbiologischer Eigenschaften.
Umgang mit Ensembl
Der Anwender wählt zunächst den ihn interessierenden Organismus. Die weitere Navigation geschieht über die Suche nach Genen oder Proteinen mit bestimmten Eigenschaften oder über die Auswahl eines Chromosoms. Von dort ist es möglich, zueinander äquivalente Regionen der Chromosomen verschiedener Organismen darzustellen.
Manchmal zeigt ein Sequenz-Vergleich, dass sich zwei Genome lokal sehr ähneln (Abbildung 2). Es muss also einen gemeinsamen Urahnen gegeben haben, etwa ein Säugetier, von dem aus sich Mensch und Maus entwickelt haben. Nur deshalb ist es möglich, Ergebnisse von Nagetier-Experimenten auf den Menschen zu übertragen. Die wichtigste Darstellungsart ist die Contig View (Abbildung 3). Ein Contig ist ein durchgängig bekannter Bereich auf einem Chromosom. Außerhalb der Contigs liegen unsequenzierte Bereiche oder solche, deren Sequenz noch nicht häufig genug bestätigt wurde.
Von der Contig View ausgehend finden die Forscher auch Verweise auf Informationen in anderen Datenbanken, die beispielsweise die molekularen Ursachen von Krankheiten beschreiben oder Proteine funktionell und strukturell klassifizieren. Daten lassen sich hier aus dem System heraus exportieren, entweder als Bild oder sogar in Form von Excel-Tabellen. Die Sequenzierung des Genoms liefert DNA-Sequenzen ohne eine weitere Beschreibung. Nur ein sehr kleiner Bruchteil der DNA-Sequenz kodiert tatsächlich Proteine.
|
Genabschnitte und |
|---|
|
Ensembl kann dabei helfen, genetisch bedingten Krankheiten auf die Spur zu kommen. Es liefert aber lediglich Hypothesen. Für Beweise sind immer Laborversuche und klinische Forschung notwendig. Manche Krankheiten gehen mit einer Verschiebung von Stücken eines Chromosoms einher, einer so genannten Translokation. Sie ist unter dem Mikroskop sichtbar. So weiß man beispielsweise[12], dass mitunter bei MDS (Myelodysplastisches Syndrom), einer Vorform einer Leukämie (Blutkrebs), ein Stück des Chromosoms 11 auf das Chromosom 21 verschoben ist. Es ist aber schwierig herauszufinden, welche Gene davon betroffen sind und weshalb dies Krebs begünstigt. Auch bleibt im Unklaren, ob es Zusammenhänge mit anderen Forschungsergebnissen zu MDS gibt. Hier macht sich Ensembl nützlich. Es erlaubt Gene einerseits danach auszuwählen, wo sie sich auf welchem Chromosom befinden. Andererseits ist es möglich, nach anderen Merkmalen zu selektieren, beispielsweise nach einem schon bekannten Zusammenhang mit Krankheiten[13]. Beispielfall LeukämieDie Suche mit dem Stichwort »Myelodysplastic syndrome« liefert zwar Gene zurück, diese liegen aber nicht auf den betroffenen Chromosomen 11 oder 21. Die relevanten Regionen dort müssen daher mit der Contig View genauer durchforstet werden. Tatsächlich entdeckt man in Region 11q24 das Gen HSPA8, von dem bekannt ist, dass es Wachstumsgene reguliert. Ungebremstes Wachstum ist es ja gerade, was einen Krebs ausmacht. Eine erste Hypothese geht davon aus, dass die Translokation die Ausleserate dieses Gens verändert und somit eine ausreichende Kontrolle des Zellwachstums erschwert. Auch Chromosom 21 liefert Hinweise. In der Region 21q11.2 fallen die Gene NRIP1 und SAMSN1 auf, deren Verantwortlichkeit für andere Krebsarten bereits nachgewiesen wurde. Ensembl zeigt zudem, dass in dieser Region auf Chromosom 21 Gene liegen oder liegen könnten, die zuvor noch niemandem aufgefallen sind (novel). Wenn man diese gezielt untersucht, besteht eine gewisse Chance, neue therapeutische Ansätze zu finden. |
Unbekannte Welten
Der übrige Bereich ist auf verschiedenste und meist noch unverstandene Weisen für die Regulierung der Gene selbst verantwortlich oder es sind nicht-ausgelesene Überbleibsel der evolutionären Entwicklung. Die Bestimmung von kodierenden Bereichen, also den Genen, ist von besonders großem Interesse und folglich ein Forschungsschwerpunkt der Ensembl-Gründer. Hierzu werden mehrere Algorithmen miteinander kombiniert[11]. Von diesen arbeiten einige ausschließlich mit der DNA-Sequenz, andere nutzen auch andere Datenquellen, beispielsweise die Protein-Datenbank SWISS-PROT[8].
So gut und schnell das Team der Ensembl-Entwickler auch sein mag, es gibt immer irgend etwas, das eine Forschungsgruppe unbedingt und sofort implementiert haben möchte. Dank offener Quellen und der Verwendung weit verbreiteter Technologien ist das kein Problem. In Rostock entsteht auf diese Weise zum Beispiel eine Erweiterung, die dem besseren Verständnis von Erbkrankeiten dient.
Auch für die Industrie ist Ensembl eine sehr nützliche Wissensquelle. Die lokale Installation von Ensembl im LAN der Forschungseinrichtung erlaubt sowohl den Schutz geistigen Eigentums als auch die Integration mit vielen internen Datenbanken. Dafür gibt es bereits eine Schnittstelle zum verteilten Annotations-System DAS[10]. Oft ist aber auch hier eine Erweiterung des Quellcodes erforderlich.
Die MySQL-Datenbanken von Ensembl sind recht groß, in Rostock belegen sie beispielsweise 65 GByte. Und es steht fest, dass die Datenmenge selbst dann weiter anwachsen wird, wenn keine neuen Organismen hinzukommen – die Beschreibung der bereits eingetragenen Genome wird ständig besser.
Es gibt leider bislang keinen Mechanismus, der ein partielles Update von Daten ermöglicht. Dies ist schwierig zu realisieren, da nicht nur die Datenmenge wächst, sonders sich oft auch das Datenbankschema verändert. Allerdings bietet die Ensembl-Gruppe einen MySQL-Server zur öffentlichen Nutzung über das Internet an – für alle, die ohne Updates der lokalen Daten auf dem neuesten Stand bleiben wollen.
Die Dokumentation ist bei Projekten dieser Art, in denen Informatiker und Nicht-Informatiker eng zusammenarbeiten, natürlich besonders wichtig. Die Online-Hilfe für Ensembl-Anwender ist sehr gut gelungen und allen, die das System lokal erweitern müssen, steht das Entwicklerteam über die Mailingliste hilfreich zur Seite.
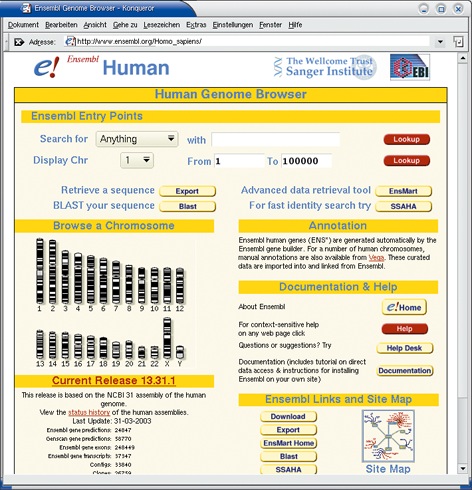
Abbildung 1: Die Ensembl-Einstiegsseite für das menschliche Genom. In der Mitte links der halbe Chromosomensatz des Menschen, der Karyotyp. Mit einem Mausklick auf ein Chromosom beginnt der Nutzer durch das Genom zu browsen.
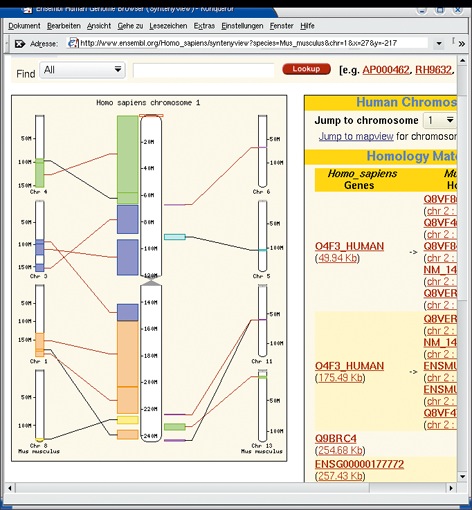
Abbildung 2: Die Chromosomen von Mensch und Maus haben zueinander äquivalente Regionen. Ensembl erlaubt jederzeit direkte Vergleiche.
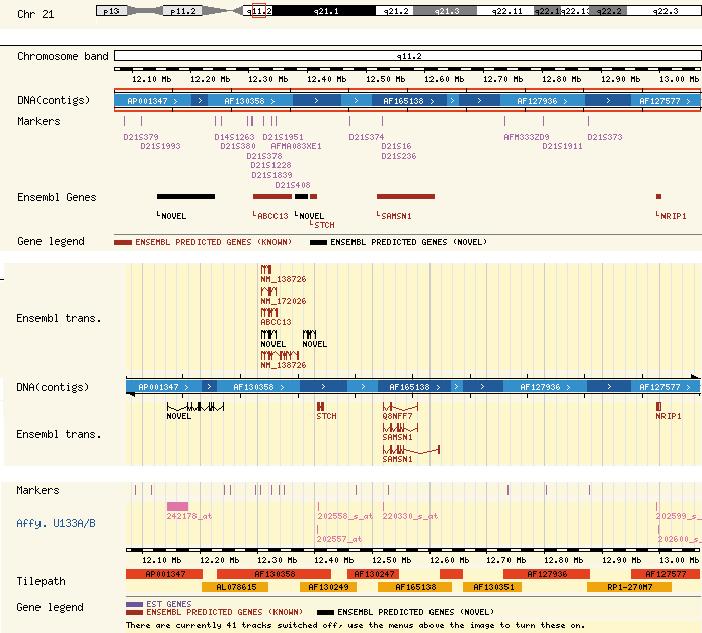
Abbildung 3: Ausschnitt aus der Contig View. So sind Gene auf den Chromosomen lokalisiert.
Fazit
Die Verfügbarkeit des Quellcodes macht Ensembl wertvoll für jede Bioinformatik-Serviceabteilung und viele Forschungsgruppen. Das System stellt nun seit mehr als zwei Jahren viele GByte Daten zu Verfügung. Dabei hat sich auch MySQL als robustes Werkzeug erwiesen. Ein aktueller günstiger Rechner mit 256 MByte Hauptspeicher, 50 GByte Festplattenkapazität und Linux reicht für den lokalen Betrieb aus. Die MySQL-Datenbank allein für das menschliche Genom ist 16 GByte groß, das der Maus belegt derzeit 8 GByte.
Für den Biologie-Unterricht, den ambitionierten Hobbyisten oder interessierten Teilnehmern an “Jugend forscht”[4] stehen damit alle Türen offen, sich mit den modernen Methoden der Genetik vertraut zu machen. Außerdem sollten sich ernsthaft Interessierte nicht scheuen, bei der nächstgelegenen Universität anzuklopfen. (uwo)
|
Infos |
|---|
|
[1] Ensembl: [http://www.ensembl.org] [2] Europäisches Molekularbiologie-Labor: [http://www.embl-heidelberg.de] [3] Europäisches Bioinformatik Institut: [http://www.ebi.ac.uk] [4] Jugend forscht:[http://www.jugend-forscht.de] [5] MDS-Studie: [http://www.gpoh.de/Studien/MDS.html] [6] Sanger Center:[http://www.sanger.ac.uk] [7] Wiki: [http://www.wiki.org] [8] Proteindatenbank SWISS-PROT: [http://www.expasy.org/sprot/] [9] BMBF-Leitprojekt “Proteom-Analyse des Menschen”: [http://www.proteome-alliance.de] [10] DAS-Schnittstelle: [http://www.biodas.org] [11] Genewise-Algorithmen: [http://www.cgen.com/products/genewise.htm] [12] MDS-Ursachen: [http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=retrieve&db =PubMed&list_uids=10451699&dopt=Abstract] [13] Online Mendelian Inheritance of Man: [http://www.ncbi.nlm.nih.gov] [14] Genome Monitoring Table: [http://www2.ebi.ac.uk/Genomsmot/] [15] Sanger Center Press Release: [http://www.sanger.ac.uk/Info/Press/2003/030414.shtml] |
|
Die |
|---|
|
Steffen Möller ist wissenschaftlicher Mitarbeiter am Proteom-Zentrum der Universität Rostock für das BMBF Projekt “Proteom-Analyse des Menschen”. Luca Toldo entwickelt und forscht in der Abteilung Scientific Information Systems/ Biological Services für die Firma Merck KGaA in Darmstadt. Die Autoren bedanken sich bei Pablo Serrano-Fernandez und Daniel Bayer für ihre konstruktiven Anmerkungen zum Artikel. |





